Eine wesentliche Gemeinsamkeit aller Unterformen der familiären partiellen Lipodystrophie ist der Verlust von subkutanem Fettgewebe an den unteren Extremitäten und am Gesäß. Dieser beginnt meist in der Pubertät.
Eine wesentliche Gemeinsamkeit aller Unterformen der familiären partiellen Lipodystrophie ist der Verlust von subkutanem Fettgewebe an den unteren Extremitäten und am Gesäß. Dieser beginnt meist in der Pubertät.
Bei der familiären partiellen Lipodystrophie Typ 1 kommt es zu einer vermehrten Fettgewebsanlagerung im Gesicht, am Hals und abdominell.
Bei der familiären partiellen Lipodystrophie Typ 2 sind neben Beinen und Gesäß auch die Arme vom Fettgewebsverlust betroffen. Es treten vermehrten Fettgewebsanlagerung in Gesicht und am Hals auf, so dass ein cushingoider Habitus resultiert.
Bei der familiären partiellen Lipodystrophie Typ 3 ist das subkutane Fettgewebe im Gesicht und am Hals normal ausgeprägt.
Verschiedene Gene wurden als ursächlich für die einzelnen Unterformen der Familiären partiellen Lipodystrophie identifiziert. Der Erbgang ist meist autosomal-dominant.
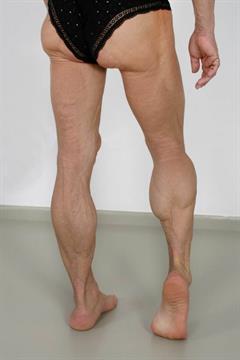 |  |
Muskulöses Aussehen und Hervortreten der Venenzeichnung durch fehlendes subkutanes Fettgewebe an Armen und Beinen bei Familiärer partieller Lipodystrophie Typ 2 |